
Latest Trends in Clinically Relevant Dissolution Specifications
Seda’s Paul Dickinson is contributing author on a recent publication regarding clinically relevant dissolution specifications (CRDS) for oral drug products.
The publication, Conference Report Developing Clinically Relevant Dissolution Specifications (CRDSs) for Oral Drug Products: Virtual Webinar Series was published earlier this month in Pharmaceutics as a collaborative effort between an eminent group of authors from across industry, academia, and regulatory bodies. The report is the output from a webinar series that was organised by the Academy of Pharmaceutical Sciences Biopharmaceutics focus group in 2021. The six-webinar series focused on the challenges of developing CRDSs for oral drug products, with discussions on progress in the field, emerging trends, and areas for continued collaboration and harmonisation.
A CRDS is a specification that takes into consideration the clinical effect of variations in dissolution ensuring a consistent safety and efficacy profile. Physiologically based biopharmaceutics modelling (PBBM) can be used to develop/support a CRDS. The aim of a PBBM is to achieve a mechanistic model that describes the drug substance, drug product, and physiological properties and processes involved from the point of administration of the drug, (with particular consideration here to solid, oral dosage forms) to absorption and subsequent distribution, metabolism, and excretion.
The report found that continued progress is being made in the field of CRDSs, and the successful application of physiologically based biopharmaceutics modelling (PBBM) is on the rise. However, there remains a clear need to continue the momentum and dialogue between the industry and regulators. Five key areas were identified which require further discussion and harmonisation.
These key areas are:
- PBBM modelling approaches and their utility, including model verification and validation. Further dialogue is required to clearly understand requirements and manage expectations both for the industry and regulators;
- Role of dissolution data (QC or more biorelevant media) as appropriate input into PBBM models;
- Clinical study design to support the setting of clinically relevant dissolution specifications;
- Regulators and the industry should develop a CRDS roadmap and framework for implementing CRDSs;
- Opportunity to engage and set up an EMA – Industry workgroup on CRDSs and PBBM in pharmaceutical applications.
Under Paul’s direction, Seda are recognised leaders in the field of clinically relevant dissolution and this knowledge along with biorelevant dissolution data (which can be generated in Seda’s laboratory) allows screening and development of formulations that are most likely to produce the required exposure and optimal pharmacokinetic profiles in patients. In later phases of development and the post launch setting, clinically relevant dissolution ensures consistent clinical performance between different batches of drug product and is a critical regulatory expectation.

For a number of years Paul and other Seda staff have been active proponents of dissolution tests that are linked to clinical performance with their landmark 2008 paper Dickinson, Lee, Stott et al. 2008 coining the term ‘clinical relevance”.
Paul Dickinson shares some personal reflections on Clinically Relevant Specifications below:
Different parts of society have subtly different expectations for medicines and how this relates to meaningful / clinically relevant specification (unambiguous test specification) (Table 1).
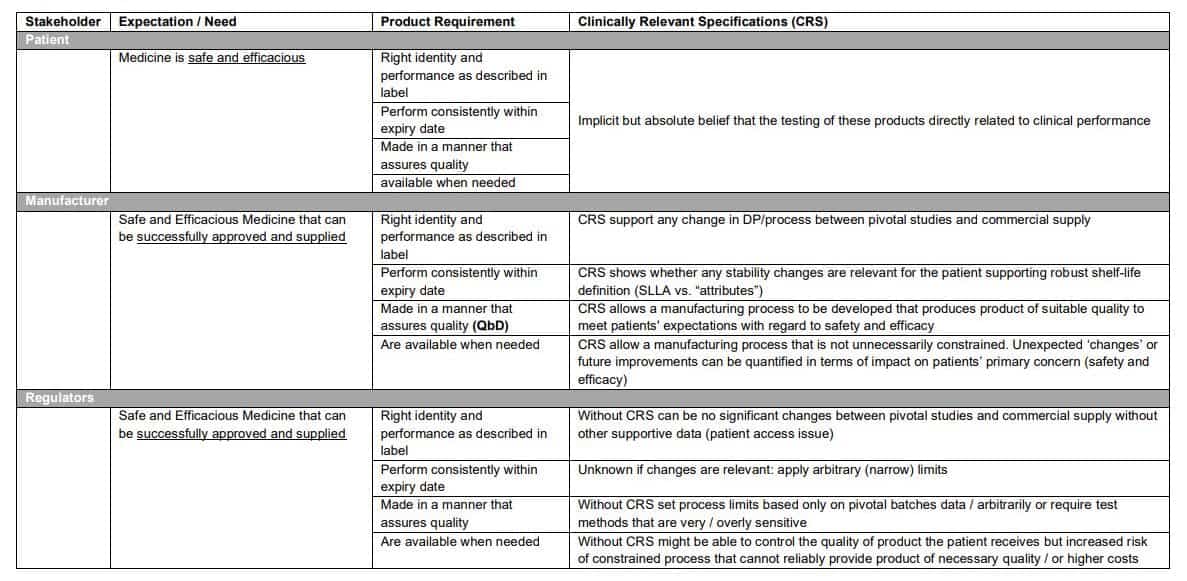
Some Clinically Relevant Specifications are uncontroversial and well accepted such as:
- Identity (Safety and Efficacy)
- Assay (Safety and Efficacy)
- Related Substances/Degradants (Safety)
For these unambiguous specifications and methods can be developed that can accurately measure these attributes and different methods (such as a different HPLC column and gradient) will give the ‘same’ result if suitably developed. For example, related substances meet ICH limits or have been qualified in toxicology / safety studies.
Against this background clinically relevant specification appear uncontroversial so why is dissolution testing so challenging?
This is related to two different expectations for dissolution testing as noted in significant regulatory guidances:
Dissolution testing as an important surrogate of clinical performance: “Drug absorption from a solid dosage form after oral administration depends on the release of the drug substance from the drug product, the dissolution or solubilization of the drug under physiological conditions, and the permeability across the gastrointestinal tract. Because of the critical nature of the first two of these steps, in vitro dissolution may be relevant to the prediction of in vivo performance. Based on this general consideration, in vitro dissolution tests for immediate release solid oral dosage forms, such as tablets and capsules, are used to (1) assess the lot-to-lot quality of a drug product; (2) guide development of new formulations; and (3) ensure continuing product quality and performance after certain changes, such as changes in the formulation, the manufacturing process, the site of manufacture, and the scale-up of the manufacturing process”.
Routine test of product quality: “A dissolution procedure intended to be used as a routine control test for immediate release drug products should be robust, reproducible and discriminatory in order to assure a consistent product quality and to detect product quality attributes, which, if altered, may affect the in vivo performance”.
This mixed role (in vivo performance and quality testing) leads to the challenges experienced in defining / accepting a clinically relevant dissolution specification. Particularly when considering quality aspects, the dissolution test is seen as a ‘global’ test that may be able to monitor both product attributes and variability in many aspects of the input materials and manufacturing process such as:
- Formulation Composition (Product attributes)
- Solubility of Drug Particles (Product attributes)
- Erosion / Disintegration of the dosage form (Product attributes)
- Granule Porosity (Process linked product attributes)
- Granule Density (Process linked product attributes)
- Tablet Hardness (Process linked product attributes)
- Granule Surface Area (Process linked product attributes)
- Drug Form & Particle Size (Process linked product attributes)
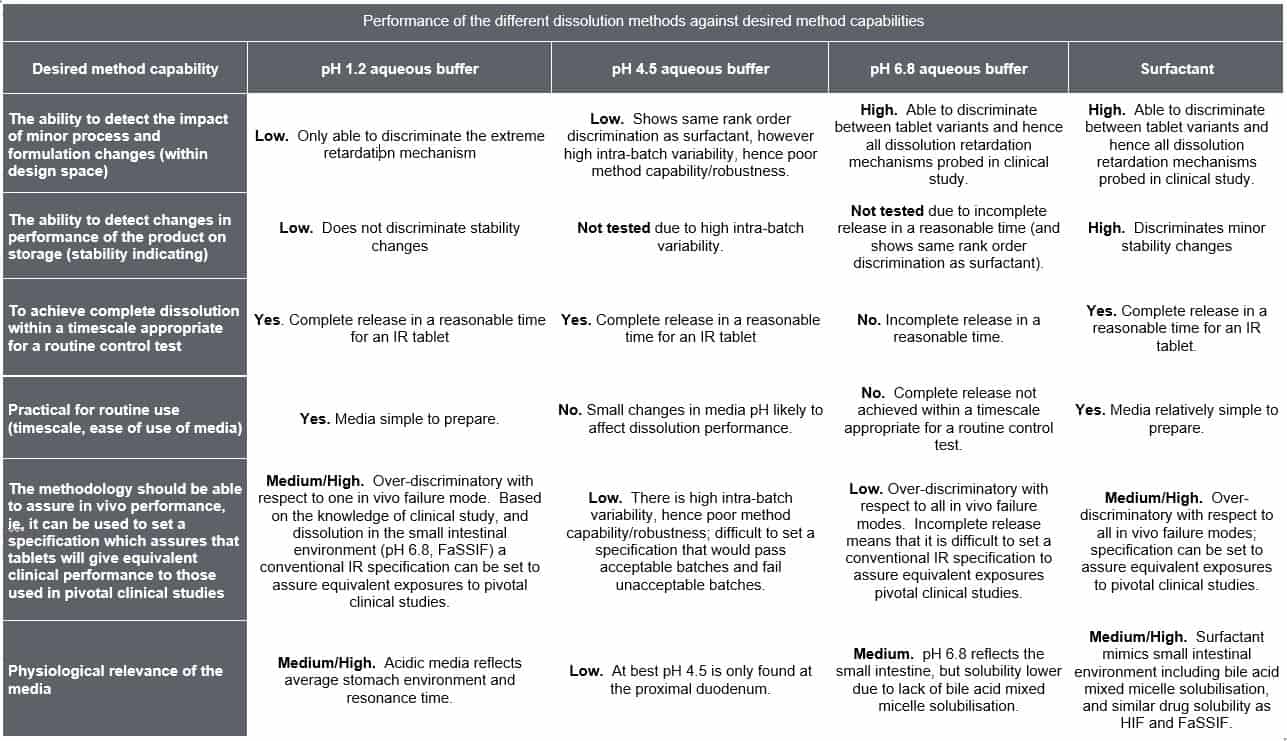
Based on these demands the dissolution test then has a number of desired capabilities (See Column 1, Table 2) and unlike an appropriately validated assay and related substance method it is possible that different dissolution methods can meet the individual method capability requirements differently (Table 1). Depending on the background of the individual considering the dissolution method they may also weight the method capabilities differently and therefore which method is most appropriate. This is a key challenge for CRDS. Although the focus of CRDS is generally on oral drug products these concepts apply equally drug products where there is a release or dissolution step.
When first considering CRDS superficially it can seem to be all about clinical studies on drug product variants with different dissolution profiles and the dissolution test. However, CRDS should be viewed as the end product of efforts to build a knowledge base which allows the understanding of clinical performance to be developed and to then allow the level of risk associated with the test and control of clinical quality to be described such that the performance (quality) of product delivered to the patient is acceptable and thus involves many disciplines.
Thus, at Seda, we believe that a systems approach to developing the understanding required to underpin CRDS was required and that good biopharmaceutics risk understanding / ability to describe clinical performance is required. This requires that this is thought about right from the start of clinical development (CRDS is usually seen as a later phase activity). It was suggested that BioRAM is a useful framework to implement systems thinking throughout development and highlights the concept of an early QTPP. BioRAM is focused on answering critical questions and moving quickly to decision point to ensures that patient centric drug products are developed and the necessary knowledge and data exists to ensure appropriate drug product quality can be maintained in pivotal studies and on and after launch. The focus of BioRAM is patient centric product design and the roadmap including early QTPP plus scoring grid facilitate this process and will result in CRDS.
Pharmaceutics is a peer-reviewed, open access journal on the science and technology of pharmaceutics and biopharmaceutics, and is published monthly online by MDPI.
For a list of other publications by Seda authors please visit Publications | SEDA (sedapds.com).
The full text, open access report can be accessed here: https://doi.org/10.3390/pharmaceutics14051010
References:
- P.A Dickinson, W.W. Lee, P.W. Stott, A.I. Townsend, J.P. Smart, P. Ghahramani, T. Hammett, L. Billett, S. Behn, R.C. Gibb and B. Abrahamsson (2008) Clinical Relevance of Dissolution Testing in Quality by Design. AAPS J. 10:380-390, http://dx.doi.org/10.1208/s12248-008-9034-7
- Guidance for Industry Dissolution Testing of Immediate Release Solid Oral Dosage Forms, 1997; U.S. Department of Health and Human Services Food and Drug Administration (CDER)
- Reflection paper on the dissolution specification for generic solid oral immediate release products with systemic action, EMA/CHMP/CVMP/QWP/336031/2017
- A. Selen, P.A. Dickinson, A. Müllertz, J.R. Crison, H.B. Mistry, M.T. Cruañes, M.N. Martinez, H. Lennernäs, T.L. Wigal, D.C. Swinney, J.E. Polli, A.T.M. Serajuddin, J.A. Cook, J.B. Dressman (2014) The Biopharmaceutics Risk Assessment Roadmap for Optimizing Clinical Drug Product Performance. J. Pharm Sci. 103: 3377–3397. http://dx.doi.org/10.1002/jps.24162
- P.A. Dickinson, F. Kesisoglou, T. Flanagan, M.N. Martinez, H.B. Mistry, J.R. Crison, J.E. Polli, M.T. Cruañes, A.T.M. Serajuddin, A. Müllertz, J.A. Cook and A. Selen (2016) Optimizing Clinical Drug Product Performance: Applying Biopharmaceutics Risk Assessment Roadmap (BioRAM) and the BioRAM Scoring Grid. J. Pharm. Sci. 105: 3243-3255. http://dx.doi.org/10.1016/j.xphs.2016.07.024
- A. Selen, A. Müllertz, F. Kesisoglou, R.J.Y. Ho, J.A. Cook, P.A. Dickinson and T. Flanagan (2020) Integrated Multi-stakeholder Systems Thinking Strategy: Decision-making with Biopharmaceutics Risk Assessment Roadmap (BioRAM) to Optimize Clinical Performance of Drug Products. AAPS J 22: 97- 117. https://doi.org/10.1208/s12248-020-00470-z